Antisense RNA
Messenger RNA (mRNA) is single-stranded. Its sequence of nucleotides is called "sense" because it results in a gene product (protein). Normally, its unpaired nucleotides are "read" by transfer RNA anticodons as the ribosome proceeds to translate the message. (See mechanism of translation.)

However, RNA can form duplexes just as DNA does. All that is needed is a second strand of RNA whose sequence of bases is complementary to the first strand; e.g.,
5´ C A U G 3´ mRNA
3´ G U A C 5´ Antisense RNA
3´ G U A C 5´ Antisense RNA
The second strand is called the antisense strand because its sequence of nucleotides is the complement of message sense. When mRNA forms a duplex with a complementary antisense RNA sequence, translation is blocked.This may occur because 
With recombinant DNA methods, synthetic genes (DNA) encoding antisense RNA molecules can be introduced into the organism.
Example: the Flavr Savr tomato
Most tomatoes that have to be shipped to market are harvested before they are ripe. Otherwise, ethylene synthesized by the tomato causes them to ripen and spoil before they reach the customer.
Transgenic tomatoes have been constructed that carry in their genome an artificial gene (DNA) that is transcribed into an antisense RNA complementary to the mRNA for an enzyme involved in ethylene production. These tomatoes make only 10% of the normal amount of the enzyme.
The goal of this work was to provide supermarket tomatoes with something closer to the appearance and taste of tomatoes harvested when ripe. However, these tomatoes often became damaged during shipment and handling and have been taken off the market.
Amplified fragment length polymorphism
AFLP-PCR or just AFLP is a PCR-based tool used in genetics research, DNA fingerprinting, and in the practice of genetic engineering. Developed in the early 1990s by Keygene,[1] AFLP uses restriction enzymes to digest genomic DNA, followed by ligation of adaptors to the sticky ends of the restriction fragments. A subset of the restriction fragments is then selected to be amplified. This selection is achieved by using primers complementary to the adaptor sequence, the restriction site sequence and a few nucleotides inside the restriction site fragments (as described in detail below). The amplified fragments are separated and visualized on denaturing polyacrylamide gels, either through autoradiography or fluorescence methodologies, or via automated capillary sequencing instruments.
AFLP is not an acronym and, despite hundreds of publications that do so, it is incorrect to refer to AFLP as "Amplified fragment length polymorphism",[2] as the resulting data are not scored as length polymorphisms, but instead as presence-absence polymorphisms.
AFLP-PCR is a highly sensitive method for detecting polymorphisms in DNA. The technique was originally described by Vos and Zabeau in 1993.[3][4] In detail, the procedure of this technique is divided into three steps:[5]
- Digestion of total cellular DNA with one or more restriction enzymes and ligation of restriction half-site specific adaptors to all restriction fragments.
- Selective amplification of some of these fragments with two PCR primers that have corresponding adaptor and restriction site specific sequences.
- Electrophoretic separation of amplicons on a gel matrix, followed by visualisation of the band pattern.
A variation on AFLP is cDNA-AFLP[6], which is used to quantify differences in gene expression levels.
Another variation on AFLP is TE Display, used to detect transposable element mobility.
Applications
The AFLP technology has the capability to detect various polymorphisms in different genomic regions simultaneously. It is also highly sensitive and reproducible. As a result, AFLP has become widely used for the identification of genetic variation in strains or closely related species of plants, fungi, animals, and bacteria. The AFLP technology has been used in criminal and paternity tests, also to determine slight differences within populations, and in linkage studies to generate maps for quantitative trait locus (QTL) analysis.
There are many advantages to AFLP when compared to other marker technologies including randomly amplified polymorphic DNA (RAPD), restriction fragment length polymorphism (RFLP), and microsatellites. AFLP not only has higher reproducibility, resolution, and sensitivity at the whole genome level compared to other techniques,[7] but it also has the capability to amplify between 50 and 100 fragments at one time. In addition, no prior sequence information is needed for amplification (Meudt & Clarke 2007).[8] As a result, AFLP has become extremely beneficial in the study of taxa including bacteria, fungi, and plants, where much is still unknown about the genomic makeup of various organisms.
Restriction fragment length polymorphism
In molecular biology, restriction fragment length polymorphism, or RFLP (commonly pronounced “rif-lip”), is a technique that exploits variations in homologous DNA sequences. It refers to a difference between samples of homologous DNA molecules that come from differing locations of restriction enzyme sites, and to a related laboratory technique by which these segments can be illustrated. In RFLP analysis, the DNA sample is broken into pieces (digested) by restriction enzymes and the resulting restriction fragments are separated according to their lengths by gel electrophoresis. Although now largely obsolete due to the rise of inexpensive DNA sequencing technologies, RFLP analysis was the first DNA profiling technique inexpensive enough to see widespread application. In addition to genetic fingerprinting, RFLP was an important tool in genome mapping, localization of genes for genetic disorders, determination of risk for disease, and paternity testing.
Analysis technique
The basic technique for detecting RFLPs involves fragmenting a sample of DNA by a restriction enzyme, which can recognize and cut DNA wherever a specific short sequence occurs, in a process known as a restriction digest. The resulting DNA fragments are then separated by length through a process known as agarose gel electrophoresis, and transferred to a membrane via the Southern blot procedure. Hybridization of the membrane to a labeled DNA probe then determines the length of the fragments which are complementary to the probe. An RFLP occurs when the length of a detected fragment varies between individuals. Each fragment length is considered an allele, and can be used in genetic analysis.
RFLP analysis may be subdivided into single- (SLP) and multi-locus probe (MLP) paradigms. Usually, the SLP method is preferred over MLP because it is more sensitive, easier to interpret and capable of analyzing mixed-DNA samples.[citation needed] Moreover, data can be generated even when the DNA is degraded (e.g. when it is found in bone remains.)
[edit] Examples
There are two common mechanisms by which the size of a particular restriction fragment can vary. In the first schematic, a small segment of the genome is being detected by a DNA probe (thicker line). In allele "A", the genome is cleaved by a restriction enzyme at three nearby sites (triangles), but only the rightmost fragment will be detected by the probe. In allele "a", restriction site 2 has been lost by a mutation, so the probe now detects the larger fused fragment running from sites 1 to 3. The second diagram shows how this fragment size variation would look on a Southern blot, and how each allele (two per individual) might be inherited in members of a family.
In the third schematic, the probe and restriction enzyme are chosen to detect a region of the genome that includes a variable VNTR segment (boxes). In allele "c" there are five repeats in the VNTR, and the probe detects a longer fragment between the two restriction sites. In allele "d" there are only two repeats in the VNTR, so the probe detects a shorter fragment between the same two restriction sites. Other genetic processes, such as insertions, deletions, translocations, and inversions, can also lead to RFLPs.
[edit] Applications
Analysis of RFLP variation in genomes was a vital tool in genome mapping and genetic disease analysis. If researchers were trying to initially determine the chromosomal location of a particular disease gene, they would analyze the DNA of members of a family afflicted by the disease, and look for RFLP alleles that show a similar pattern of inheritance as that of the disease (see Genetic linkage). Once a disease gene was localized, RFLP analysis of other families could reveal who was at risk for the disease, or who was likely to be a carrier of the mutant genes.
RFLP analysis was also the basis for early methods of Genetic fingerprinting, useful in the identification of samples retrieved from crime scenes, in the determination of paternity, and in the characterization of genetic diversity or breeding patterns in animal populations.
Alkaline phosphatase
Alkaline phosphatase (ALP, ALKP) (EC 3.1.3.1) is a hydrolase enzyme responsible for removing phosphate groups from many types of molecules, including nucleotides, proteins, and alkaloids. The process of removing the phosphate group is called dephosphorylation. As the name suggests, alkaline phosphatases are most effective in an alkaline environment. It is sometimes used synonymously as basic phosphatase.[2]
Use in research
Typical use in the lab for alkaline phosphatases includes removing phosphate monoester to prevent self ligation.[8]
Common alkaline phosphatases used in research include:
- Placental alkaline phosphatase (PALP) and its C terminally truncated version that lacks the last 24 amino acids (constituting the domain that targets for GPI membrane anchoring) - the secreted alkaline phosphatase (SEAP)
Alkaline phosphatase has become a useful tool in molecular biology laboratories, since DNA normally possesses phosphate groups on the 5' end. Removing these phosphates prevents the DNA from ligating (the 5' end attaching to the 3' end), thereby keeping DNA molecules linear until the next step of the process for which they are being prepared; also, removal of the phosphate groups allows radiolabeling (replacement by radioactive phosphate groups) in order to measure the presence of the labeled DNA through further steps in the process or experiment. For these purposes, the alkaline phosphatase from shrimp is the most useful, as it is the easiest to inactivate once it has done its job.
Undifferentiated pluripotent stem cells have elevated levels of alkaline phosphatase on their cell membrane, therefore alkaline phosphatase staining is used to detect these cells and to test pluripotency (i.e. embryonic stem cells or embryonal carcinoma cells).[9]
One common use in the dairy industry is as a marker of pasteurisation in cows' milk. This molecule is denatured by elevated temperatures found during pasteurisation, and can be tested for via colour change of a para-Nitrophenylphosphate substrate in a buffered solution (Aschaffenburg Mullen Test).[10] Raw milk would typically produce a yellow colouration within a couple of minutes, whereas properly pasteurised milk should show no change. There are of course exceptions to this in the case of heat-stable alkaline phophatases produced by some bacteria.
DNA ligase
In molecular biology, DNA ligase is a specific type of enzyme, a ligase, (EC 6.5.1.1) that facilitates the joining of DNA strands together by catalyzing the formation of a phosphodiester bond. It plays a role in repairing single-strand breaks in duplex DNA in living organisms, but some forms (such as DNA ligase IV) may specifically repair double-strand breaks (i.e. a break in both complementary strands of DNA). Single-strand breaks are repaired by DNA ligase using the complementary strand of the double helix as a template,[1] with DNA ligase creating the final phosphodiester bond to fully repair the DNA.
DNA ligase has applications in both DNA repair and DNA replication (see Mammalian ligases). In addition, DNA ligase has extensive use in molecular biology laboratories for genetic recombination experiments (see Applications in molecular biology research). Purified DNA ligase is used in gene cloning to join DNA molecules together to form recombinant DNA.
Ligase mechanism
The mechanism of DNA ligase is to form two covalent phosphodiester bonds between 3' hydroxyl ends of one nucleotide, ("acceptor") with the 5' phosphate end of another ("donor"). ATP is required for the ligase reaction, which proceeds in three steps:
- adenylation (addition of AMP) of a residue in the active center of the enzyme, pyrophosphate is released;
- transfer of the AMP to the 5' phosphate of the so-called donor, formation of a pyrophosphate bond;
- formation of a phosphodiester bond between the 5' phosphate of the donor and the 3' hydroxyl of the acceptor.[2]

Ligase will also work with blunt ends, although higher enzyme concentrations and different reaction conditions are required.
[edit] Applications in molecular biology research
DNA ligases have become an indispensable tool in modern molecular biology research for generating recombinant DNA sequences. For example, DNA ligases are used with restriction enzymes to insert DNA fragments, often genes, into plasmids.
One vital aspect to performing efficient recombination experiments involving the ligation of cohesive-ended fragments is controlling the optimal temperature. Most experiments use T4 DNA Ligase (isolated from bacteriophage T4), which is most active at 25°C. However, for optimal ligation efficiency with cohesive-ended fragments ("sticky ends"), the optimal enzyme temperature needs to be balanced with the melting temperature Tm (also the annealing temperature) of the sticky ends being ligated.[4] If the ambient temperature exceeds Tm, the homologous pairing of the sticky ends would not be stable because the high temperature disrupts hydrogen bonding. Ligation reaction is most efficient when the sticky ends are already stably annealed, disruption of the annealing ends would therefore results in low ligation efficiency. The shorter the overhang, the lower the Tm, typically a 4-base overhang has a Tm of 12-16°C.
Since blunt-ended DNA fragments have no cohesive ends to anneal, the melting temperature is not a factor to consider within the normal temperature range of the ligation reaction. However, the higher the temperature, the less chance that the ends to be joined will be aligned to allow ligation (molecules move around the solution more at higher temperatures). The limiting factor in blunt end ligation is not the activity of the ligase but rather the number of alignments between DNA fragment ends that occur. The most efficient ligation temperature for blunt-ended DNA would therefore be the temperature at which the greatest number of alignments can occur. Therefore, the majority of blunt-ended ligations are carried out at 14-20°C overnight. The absence of a stably annealed ends also means that the ligation efficiency is lowered, requiring a higher ligase concentration to be used.(T4 DNA ligase is the only commercially-available DNA ligase to anneal blunt ends).[4]
RAPD
RAPD (pronounced "rapid") stands for random amplification of polymorphic DNA. It is a type of PCR reaction, but the segments of DNA that are amplified are random. The scientist performing RAPD creates several arbitrary, short primers (8–12 nucleotides), then proceeds with the PCR using a large template of genomic DNA, hoping that fragments will amplify. By resolving the resulting patterns, a semi-unique profile can be gleaned from a RAPD reaction.
No knowledge of the DNA sequence for the targeted genome is required, as the primers will bind somewhere in the sequence, but it is not certain exactly where. This makes the method popular for comparing the DNA of biological systems that have not had the attention of the scientific community, or in a system in which relatively few DNA sequences are compared (it is not suitable for forming a DNA databank). Because it relies on a large, intact DNA template sequence, it has some limitations in the use of degraded DNA samples. Its resolving power is much lower than targeted, species specific DNA comparison methods, such as short tandem repeats. In recent years, RAPD has been used to characterize, and trace, the phylogeny of diverse plant and animal species.
How it works
Unlike traditional PCR analysis, RAPD does not require any specific knowledge of the DNA sequence of the target organism: the identical 10-mer primers will or will not amplify a segment of DNA, depending on positions that are complementary to the primers' sequence. For example, no fragment is produced if primers annealed too far apart or 3' ends of the primers are not facing each other. Therefore, if a mutation has occurred in the template DNA at the site that was previously complementary to the primer, a PCR product will not be produced, resulting in a different pattern of amplified DNA segments on the gel.
[edit] Example
RAPD is an inexpensive yet powerful typing method for many bacterial species. The image visible at the link [1] is a silver-stained polyacrylamide gel showing three distinct RAPD profiles generated by primer OPE15 for Haemophilus ducreyi isolates from Tanzania, Senegal, Thailand, Europe, and North America.
![[1]](http://www.ncbi.nlm.nih.gov/projects/genome/probe/IMG/zjm0010661110001.jpg){kind=link}
Selecting the right sequence for the primer is very important because different sequences will produce different band patterns and possibly allow for a more specific recognition of individual strains.
Limitations of RAPD
- Nearly all RAPD markers are dominant, i.e. it is not possible to distinguish whether a DNA segment is amplified from a locus that is heterozygous (1 copy) or homozygous (2 copies). Codominant RAPD markers, observed as different-sized DNA segments amplified from the same locus, are detected only rarely.
- PCR is an enzymatic reaction, therefore the quality and concentration of template DNA, concentrations of PCR components, and the PCR cycling conditions may greatly influence the outcome. Thus, the RAPD technique is notoriously laboratory dependent and needs carefully developed laboratory protocols to be reproducible.
- Mismatches between the primer and the template may result in the total absence of PCR product as well as in a merely decreased amount of the product. Thus, the RAPD results can be difficult to interpret.
Developing locus-specific, co-dominant markers from RAPDs
- The polymorphic RAPD marker band is isolated from the gel.
- It is amplified in the PCR reaction.
- The PCR product is cloned and sequenced.
- New longer and specific primers are designed for the DNA sequence, which is called the Sequenced Characterized Amplified Region Marker (SCAR).
Vector (molecular biology)
In molecular cloning, a vector is a DNA molecule used as a vehicle to artificially carry foreign genetic material into another cell, where it can be replicated and/or expressed. A vector containing foreign DNA is termed recombinant DNA. The four major types of vectors are plasmids, viral vectors, cosmids, and artificial chromosomes. Common to all engineered vectors are an origin of replication, a multicloning site, and a selectable marker.
The vector itself is generally a DNA sequence that consists of an insert (transgene) and a larger sequence that serves as the "backbone" of the vector. The purpose of a vector which transfers genetic information to another cell is typically to isolate, multiply, or express the insert in the target cell. Vectors called expression vectors (expression constructs) specifically are for the expression of the transgene in the target cell, and generally have a promoter sequence that drives expression of the transgene. Simpler vectors called transcription vectors are only capable of being transcribed but not translated: they can be replicated in a target cell but not expressed, unlike expression vectors. Transcription vectors are used to amplify their insert.
Insertion of a vector into the target cell is usually called transformation for bacterial cells, transfection for eukaryotic cells, although insertion of a viral vector is often called transduction.
Characteristics
[edit] Plasmids
Plasmids are double-stranded generally circular DNA sequences that are capable of automatically replicating in a host cell. Plasmid vectors minimalistically consist of an origin of replication that allows for semi-independent replication of the plasmid in the host and also the transgene insert. Modern plasmids generally have many more features, notably including a "multiple cloning site" which includes nucleotide overhangs for insertion of an insert, and multiple restriction enzyme consensus sites to either side of the insert. In the case of plasmids utilized as transcription vectors, incubating bacteria with plasmids generates hundreds or thousands of copies of the vector within the bacteria in hours, and the vectors can be extracted from the bacteria, and the multiple cloning site can be cut by restriction enzymes to excise the hundredfold or thousandfold amplified insert. These plasmid transcription vectors characteristically lack crucial sequences that code for polyadenylation sequences and translation termination sequences in translated mRNAs, making protein expression from transcription vectors impossible. Plasmids may be conjugative/transmissible and non-conjugative:
- conjugative: mediate DNA transfer through conjugation and therefore spread rapidly among the bacterial cells of a population; e.g., F plasmid, many R and some col plasmids.
- nonconjugative- do not mediate DNA through conjugation, e.g., many R and col plasmids.
[edit] Viral vectors
Viral vectors are generally genetically-engineered viruses carrying modified viral DNA or RNA that has been rendered noninfectious, but still contain viral promoters and also the transgene, thus allowing for translation of the transgene through a viral promoter. However, because viral vectors frequently are lacking infectious sequences, they require helper viruses or packaging lines for large-scale transfection. Viral vectors are often designed for permanent incorporation of the insert into the host genome, and thus leave distinct genetic markers in the host genome after incorporating the transgene. For example, retroviruses leave a characteristic retroviral integration pattern after insertion that is detectable and indicates that the viral vector has incorporated into the host genome.
[edit] Transcription
Transcription is a necessary component in all vectors: the premise of a vector is to multiply the insert (although expression vectors later also drive the translation of the multiplied insert). Thus, even stable expression is determined by stable transcription, which generally depends on promoters in the vector. However, expression vectors have a variety of expression patterns: constitutive (consistent expression) or inducible (expression only under certain conditions or chemicals). This expression is based on different promoter activities, not post-transcriptional activities. Thus, these two different types of expression vectors depend on different types of promoters.
Expression
Expression vectors produce proteins through the transcription of the vector's insert followed by translation of the mRNA produced, they therefore require more components than the simpler transcription-only vectors. Expression in different host organism would require different elements, although they share similar requirements, for example a promoter for initiation of transcription, a ribosomal binding site for translation initiation, and termination signals.
[edit] Features
Modern vectors contain essential components as well as other additional features:
- Origin of replication: Necessary for the replication and maintenance of the vector in the host cell.
- Promoter: Promoters are used to drive the transcription of the vector's transgene as well as the other genes in the vector such as the antibiotic resistance gene.
- Cloning site: This may be a multiple cloning site or other features that allow for the insertion of foreign DNA into the vector.
- Genetic markers: Genetic markers for viral vectors allow for confirmation that the vector has integrated with the host genomic DNA.
- Antibiotic resistance: Vectors with antibiotic-resistance open reading frames allow for survival of cells that have taken up the vector in growth media containing antibiotics through antibiotic selection.
- Epitope: Vector contains a sequence for a specific epitope that is incorporated into the expressed protein. Allows for antibody identification of cells expressing the target protein.
- Targeting sequence: Expression vectors may include encoding for a targeting sequence in the finished protein that directs the expressed protein to a specific organelle in the cell or specific location such as the periplasmic space of bacteria.
Transformation (genetics)
In molecular biology transformation is genetic alteration of a cell resulting from the direct uptake, incorporation and expression of exogenous genetic material (exogenous DNA) from its surroundings and taken up through the cell membrane(s). Transformation occurs naturally in some species of bacteria, but it can also be effected by artificial means in other cells. For transformation to happen, bacteria must be in a state of competence, which might occur as a time-limited response to environmental conditions such as starvation and cell density. Transformation is one of three processes by which exogenous genetic material may be introduced into a bacterial cell, the other two being conjugation (transfer of genetic material between two bacterial cells in direct contact) and transduction (injection of foreign DNA by a bacteriophage virus into the host bacterium). "Transformation" may also be used to describe the insertion of new genetic material into nonbacterial cells, including animal and plant cells; however, because "transformation" has a special meaning in relation to animal cells, indicating progression to a cancerous state, the term should be avoided for animal cells when describing introduction of exogenous genetic material. Introduction of foreign DNA into eukaryotic cells is often called "transfection”.
Methods and mechanisms
[edit] Bacteria
Bacterial transformation may be referred to as a stable genetic change brought about by the uptake of naked DNA (DNA without associated cells or proteins) and competence refers to the state of being able to take up exogenous DNA from the environment. There are two forms of transformation and competence: natural and artificial.
Restriction enzyme
A restriction enzyme (or restriction endonuclease) is an enzyme that cuts DNA at specific recognition nucleotide sequences known as restriction sites.[1][2][3] Restriction enzymes are commonly classified into three types, which differ in their structure and whether they cut their DNA substrate at their recognition site, or if the recognition and cleavage sites are separate from one another. To cut DNA, all restriction enzymes make two incisions, once through each sugar-phosphate backbone (i.e. each strand) of the DNA double helix.
These enzymes are found in bacteria and archaea and probably evolved to provide a defense mechanism against invading viruses.[4][5] Inside a bacterium, the restriction enzymes selectively cut up foreign DNA in a process called restriction; while host DNA is protected by a modification enzyme (a methylase) that modifies the bacterial DNA and blocks cleavage. Together, these two processes form the restriction modification system.[6]
Types
Naturally occurring restriction endonucleases are categorized into four groups (Types I, II III, and IV) based on their composition and enzyme cofactor requirements, the nature of their target sequence, and the position of their DNA cleavage site relative to the target sequence.[26][27][28] All types of enzymes recognize specific short DNA sequences and carry out the endonucleolytic cleavage of DNA to give specific fragments with terminal 5'-phosphates. They differ in their recognition sequence, subunit composition, cleavage position, and cofactor requirements,[29][30] as summarised below:
- Type IV enzymes target modified DNA, e.g. methylated, hydroxymethylated and glucosyl-hydroxymethylated DNA
[edit] Type I
Type I restriction enzymes were the first to be identified and were first identified in two different strains (K-12 and B) of E. coli.[31] These enzymes cut at a site that differs, and is a random distance (at least 1000 bp) away, from their recognition site. Cleavage at these random sites follows a process of DNA translocation, which shows that these enzymes are also molecular motors. The recognition site is asymmetrical and is composed of two specific portions—one containing 3–4 nucleotides, and another containing 4–5 nucleotides—separated by a non-specific spacer of about 6–8 nucleotides. These enzymes are multifunctional and are capable of both restriction and modification activities, depending upon the methylation status of the target DNA. The cofactors S-Adenosyl methionine (AdoMet), hydrolyzed adenosine triphosphate (ATP), and magnesium (Mg2+) ions, are required for their full activity. Type I restriction enzymes possess three subunits called HsdR, HsdM, and HsdS; HsdR is required for restriction; HsdM is necessary for adding methyl groups to host DNA (methyltransferase activity) and HsdS is important for specificity of the recognition (DNA-binding) site in addition to both restriction (DNA cleavage) and modification (DNA methyltransferase) activity.[26][31]
Typical type II restriction enzymes differ from type I restriction enzymes in several ways. They are a homodimer, with recognition sites are usually undivided and palindromic and 4–8 nucleotides in length. They recognize and cleave DNA at the same site, and they do not use ATP or AdoMet for their activity—they usually require only Mg2+ as a cofactor.[23] These are the most commonly available and used restriction enzymes. In the 1990s and early 2000s, new enzymes from this family were discovered that did not follow all the classical criteria of this enzyme class, and new subfamily nomenclature was developed to divide this large family into subcategories based on deviations from typical characteristics of type II enzymes.[23] These subgroups are defined using a letter suffix.
Type IIB restriction enzymes (e.g. BcgI and BplI) are multimers, containing more than one subunit.[23] They cleave DNA on both sides of their recognition to cut out the recognition site. They require both AdoMet and Mg2+ cofactors. Type IIE restriction endonucleases (e.g. NaeI) cleave DNA following interaction with two copies of their recognition sequence.[23] One recognition site acts as the target for cleavage, while the other acts as an allosteric effector that speeds up or improves the efficiency of enzyme cleavage. Similar to type IIE enzymes, type IIF restriction endonucleases (e.g. NgoMIV) interact with two copies of their recognition sequence but cleave both sequences at the same time.[23] Type IIG restriction endonucleases (Eco57I) do have a single subunit, like classical Type II restriction enzymes, but require the cofactor AdoMet to be active.[23] Type IIM restriction endonucleases, such as DpnI, are able to recognize and cut methylated DNA.[23] Type IIS restriction endonucleases (e.g. FokI) cleave DNA at a defined distance from their non-palindromic asymmetric recognition sites.[23] These enzymes may function as dimers. Similarly, Type IIT restriction enzymes (e.g., Bpu10I and BslI) are composed of two different subunits. Some recognize palindromic sequences while others have asymmetric recognition sites.[23]
[edit] Type III
Type III restriction enzymes (e.g. EcoP15) recognize two separate non-palindromic sequences that are inversely oriented. They cut DNA about 20-30 base pairs after the recognition site.[33] These enzymes contain more than one subunit and require AdoMet and ATP cofactors for their roles in DNA methylation and restriction, respectively.[34] They are components of prokaryotic DNA restriction-modification mechanisms that protect the organism against invading foreign DNA. Type III enzymes are hetero-oligomeric, multifunctional proteins composed of two subunits, Res and Mod. The Mod subunit recognises the DNA sequence specific for the system and is a modification methyltransferase; as such it is functionally equivalent to the M and S subunits of type I restriction endonuclease. Res is required for restriction, although it has no enzymatic activity on its own.
[edit] Applications
Isolated restriction enzymes are used to manipulate DNA for different scientific applications.
They are used to assist insertion of genes into plasmid vectors during gene cloning and protein expression experiments. For optimal use, plasmids that are commonly used for gene cloning are modified to include a short polylinker sequence (called the multiple cloning site, or MCS) rich in restriction enzyme recognition sequences. This allows flexibility when inserting gene fragments into the plasmid vector; restriction sites contained naturally within genes influence the choice of endonuclease for digesting the DNA since it is necessary to avoid restriction of wanted DNA while intentionally cutting the ends of the DNA. To clone a gene fragment into a vector, both plasmid DNA and gene insert are typically cut with the same restriction enzymes, and then glued together with the assistance of an enzyme known as a DNA ligase.[48][49]
Restriction enzymes can also be used to distinguish gene alleles by specifically recognizing single base changes in DNA known as single nucleotide polymorphisms (SNPs).[50][51] This is only possible if a SNP alters the restriction site present in the allele. In this method, the restriction enzyme can be used to genotype a DNA sample without the need for expensive gene sequencing. The sample is first digested with the restriction enzyme to generate DNA fragments, and then the different sized fragments separated by gel electrophoresis. In general, alleles with correct restriction sites will generate two visible bands of DNA on the gel, and those with altered restriction sites will not be cut and will generate only a single band. The number of bands reveals the sample subject's genotype, an example of restriction mapping.[citation needed]
In a similar manner, restriction enzymes are used to digest genomic DNA for gene analysis by Southern blot. This technique allows researchers to identify how many copies (or paralogues) of a gene are present in the genome of one individual, or how many gene mutations (polymorphisms) have occurred within a population. The latter example is called restriction fragment length polymorphism (RFLP).
RAPD
RAPD (pronounced "rapid") stands for random amplification of polymorphic DNA. It is a type of PCR reaction, but the segments of DNA that are amplified are random. The scientist performing RAPD creates several arbitrary, short primers (8–12 nucleotides), then proceeds with the PCR using a large template of genomic DNA, hoping that fragments will amplify. By resolving the resulting patterns, a semi-unique profile can be gleaned from a RAPD reaction.
No knowledge of the DNA sequence for the targeted genome is required, as the primers will bind somewhere in the sequence, but it is not certain exactly where. This makes the method popular for comparing the DNA of biological systems that have not had the attention of the scientific community, or in a system in which relatively few DNA sequences are compared (it is not suitable for forming a DNA databank). Because it relies on a large, intact DNA template sequence, it has some limitations in the use of degraded DNA samples. Its resolving power is much lower than targeted, species specific DNA comparison methods, such as short tandem repeats. In recent years, RAPD has been used to characterize, and trace, the phylogeny of diverse plant and animal species.
How it works
Unlike traditional PCR analysis, RAPD does not require any specific knowledge of the DNA sequence of the target organism: the identical 10-mer primers will or will not amplify a segment of DNA, depending on positions that are complementary to the primers' sequence. For example, no fragment is produced if primers annealed too far apart or 3' ends of the primers are not facing each other. Therefore, if a mutation has occurred in the template DNA at the site that was previously complementary to the primer, a PCR product will not be produced, resulting in a different pattern of amplified DNA segments on the gel.
[edit] Example
RAPD is an inexpensive yet powerful typing method for many bacterial species. The image visible at the link [1] is a silver-stained polyacrylamide gel showing three distinct RAPD profiles generated by primer OPE15 for Haemophilus ducreyi isolates from Tanzania, Senegal, Thailand, Europe, and North America.
Selecting the right sequence for the primer is very important because different sequences will produce different band patterns and possibly allow for a more specific recognition of individual strains.
Northern blot
The northern blot is a technique used in molecular biology research to study gene expression by detection of RNA (or isolated mRNA) in a sample.[1][2]
With northern blotting it is possible to observe cellular control over structure and function by determining the particular gene expression levels during differentiation, morphogenesis, as well as abnormal or diseased conditions.[3] Northern blotting involves the use of electrophoresis to separate RNA samples by size and detection with a hybridization probe complementary to part of or the entire target sequence. The term 'northern blot' actually refers specifically to the capillary transfer of RNA from the electrophoresis gel to the blotting membrane. However, the entire process is commonly referred to as northern blotting.[4] The northern blot technique was developed in 1977 by James Alwine, David Kemp, and George Stark at Stanford University.[5] Northern blotting takes its name from its similarity to the first blotting technique, the Southern blot, named for biologist Edwin Southern.[1] The major difference is that RNA, rather than DNA, is analyzed in the northern blot.
Procedure
A general blotting procedure[4] starts with extraction of total RNA from a homogenized tissue sample or from cells. Eukaryotic mRNA can then be isolated through the use of oligo (dT) cellulose chromatography to isolate only those RNAs with a poly(A) tail.[7][8] RNA samples are then separated by gel electrophoresis. Since the gels are fragile and the probes are unable to enter the matrix, the RNA samples, now separated by size, are transferred to a nylon membrane through a capillary or vacuum blotting system.
A nylon membrane with a positive charge is the most effective for use in northern blotting since the negatively charged nucleic acids have a high affinity for them. The transfer buffer used for the blotting usually contains formamide because it lowers the annealing temperature of the probe-RNA interaction, thus preventing RNA degradation by high temperatures.[9] Once the RNA has been transferred to the membrane, it is immobilized through covalent linkage to the membrane by UV light or heat. After a probe has been labeled, it is hybridized to the RNA on the membrane. Experimental conditions that can affect the efficiency and specificity of hybridization include ionic strength, viscosity, duplex length, mismatched base pairs, and base composition.[10] The membrane is washed to ensure that the probe has bound specifically and to avoid background signals from arising. The hybrid signals are then detected by X-ray film and can be quantified by densitometry. To create controls for comparison in a northern blot, samples not displaying the gene product of interest can be used after determination by microarrays or RT-PCR.[10]
[edit] Gels
The RNA samples are most commonly separated on agarose gels containing formaldehyde as a denaturing agent for the RNA to limit secondary structure.[10][11] The gels can be stained with ethidium bromide (EtBr) and viewed under UV light to observe the quality and quantity of RNA before blotting.[10] Polyacrylamide gel electrophoeresis with urea can also be used in RNA separation but it is most commonly used for fragmented RNA or microRNAs.[12] An RNA ladder is often run alongside the samples on an electrophoresis gel to observe the size of fragments obtained but in total RNA samples the ribosomal subunits can act as size markers.[10] Since the large ribosomal subunit is 28S (approximately 5kb) and the small ribosomal subunit is 18S (approximately 2kb) two prominent bands will appear on the gel, the larger at close to twice the intensity of the smaller.[10][13]
[edit] Probes
Probes for northern blotting are composed of nucleic acids with a complementary sequence to all or part of the RNA of interest, they can be DNA, RNA, or oligonucleotides with a minimum of 25 complementary bases to the target sequence.[4] RNA probes (riboprobes) that are transcribed in vitro are able to withstand more rigorous washing steps preventing some of the background noise.[10] Commonly cDNA is created with labelled primers for the RNA sequence of interest to act as the probe in the northern blot.[14] The probes need to be labelled either with radioactive isotopes (32P) or with chemiluminescence in which alkaline phosphatase or horseradish peroxidase break down chemiluminescent substrates producing a detectable emission of light.[15] The chemiluminescent labelling can occur in two ways: either the probe is attached to the enzyme, or the probe is labelled with a ligand (e.g. biotin) for which the antibody (e.g. avidin or streptavidin) is attached to the enzyme.[10] X-ray film can detect both the radioactive and chemiluminescent signals and many researchers prefer the chemiluminescent signals because they are faster, more sensitive, and reduce the health hazards that go along with radioactive labels.[15] The same membrane can be probed up to five times without a significant loss of the target RNA.[9]
[edit] Applications
Northern blotting allows one to observe a particular gene's expression pattern between tissues, organs, developmental stages, environmental stress levels, pathogen infection, and over the course of treatment.[8][14][16] The technique has been used to show overexpression of oncogenes and downregulation of tumor-suppressor genes in cancerous cells when compared to 'normal' tissue,[10] as well as the gene expression in the rejection of transplanted organs.[17] If an upregulated gene is observed by an abundance of mRNA on the northern blot the sample can then be sequenced to determine if the gene is known to researchers or if it is a novel finding.[17] The expression patterns obtained under given conditions can provide insight into the function of that gene. Since the RNA is first separated by size, if only one probe type is used variance in the level of each band on the membrane can provide insight into the size of the product, suggesting alternative splice products of the same gene or repetitive sequence motifs.[7][13] The variance in size of a gene product can also indicate deletions or errors in transcript processing, by altering the probe target used along the known sequence it is possible to determine which region of the RNA is missing.[1]
BlotBase is an online database publishing northern blots. BlotBase has over 700 published northern blots of human and mouse samples, in over 650 genes across more than 25 different tissue types.[3] Northern blots can be searched by a blot ID, paper reference, gene identifier, or by tissue.[3] The results of a search provide the blot ID, species, tissue, gene, expression level, blot image (if available), and links to the publication that the work originated from.[3] This new database provides sharing of information between members of the science community that was not previously seen in northern blotting as it was in sequence analysis, genome determination, protein structure, etc.
[edit] Advantages and disadvantages
Analysis of gene expression can be done by several different methods including RT-PCR, RNase protection assays, microarrays, serial analysis of gene expression (SAGE), as well as northern blotting.[3][4] Microarrays are quite commonly used and are usually consistent with data obtained from northern blots; however, at times northern blotting is able to detect small changes in gene expression that microarrays cannot.[18] The advantage that microarrays have over northern blots is that thousands of genes can be visualized at a time, while northern blotting is usually looking at one or a small number of genes.[16][18]
A problem in northern blotting is often sample degradation by RNases (both endogenous to the sample and through environmental contamination), which can be avoided by proper sterilization of glassware and the use of RNase inhibitors such as DEPC (diethylpyrocarbonate).[4] The chemicals used in most northern blots can be a risk to the researcher, since formaldehyde, radioactive material, ethidium bromide, DEPC, and UV light are all harmful under certain exposures.[10] Compared to RT-PCR, northern blotting has a low sensitivity, but it also has a high specificity which is important to reduce false positive results.[10]
The advantages of using northern blotting include the detection of RNA size, the observation of alternate splice products, the use of probes with partial homology, the quality and quantity of RNA can be measured on the gel prior to blotting, and the membranes can be stored and reprobed for years after blotting.[10]
Yeast artificial chromosome
A yeast artificial chromosome (YAC) is a vector used to clone DNA fragments larger than 100 kb and up to 3000 kb. YACs are useful for the physical mapping of complex genomes and for the cloning of large genes. First described in 1983 by Murray and Szostak, a YAC is an artificially constructed chromosome that contains a centromere, telomeres and an autonomous replicating sequence (ARS) element, which are required for replication and preservation of YAC in yeast cells. ARS elements are thought to act as replication origins. A YAC is built using an initial circular plasmid, which is typically broken into two linear molecules using restriction enzymes; DNA ligase is then used to ligate a sequence or gene of interest between the two linear molecules, forming a single large linear piece of DNA.[citation needed].
A plasmid-derived origin of replication (ori) and an antibiotic resistance gene allow the YAC vector to be amplified and selected for in E. coli. TRP1 and URA3 genes are included in the YAC vector to provide a selection system for identifying transformed yeast cells that include YAC by complementing recessive alleles trp1 and ura3 in yeast host cell. YAC vector cloning site for foreign DNA is located within the SUP4 gene. This gene compensates for a mutation in the yeast host cell that causes the accumulation of red pigment. The host cells are normally red, and those transformed with YAC only, will form colourless colonies. Cloning of a foreign DNA fragment into the YAC causes insertional inactivation, restoring the red colour. Therefore the colonies that contain the foreign DNA fragment are red.[1]
Advantages and Disadvantages
Yeast expression vectors, such as YACs, YIps (yeast integrating plasmids), and YEps (yeast episomal plasmids), have an advantage over bacterial artificial chromosomes (BACs) in that they can be used to express eukaryotic proteins that require posttranslational modification.
However, YACs are significantly less stable than BACs, producing "chimeric effects": artifacts where the sequence of the cloned DNA actually corresponds not to a single genomic region but to multiple regions. Chimerism may be due to either co-ligation of multiple genomic segments into a single YAC, or recombination of two or more YACs transformed in the same host Yeast cell.[2] The incidence of chimerism may be as high as 50%.[3] Other artifacts are deletion of segments from a cloned region, and rearrangement of genomic segments (such as inversion). In all these cases, the sequence as determined from the YAC clone is different from the original, natural sequence, leading to inconsistent results and errors in interpretation if the clone's information is relied upon. Due to these issues, the Human Genome Project ultimately abandoned the use of YACs and switched to bacterial artificial chromosomes, where the incidence of these artifacts is very low.
Dot blot
A dot blot (or slot blot) is a technique in molecular biology used to detect biomolecules, and for detecting, analyzing, and identifying proteins . It represents a simplification of the northern blot, southern blot, or western blot methods. In a dot blot the biomolecules to be detected are not first separated by electrophoresis. Instead, a mixture containing the molecule to be detected is applied directly on a membrane as a dot, and then is spotted through circular templates directly onto the membrane or paper substrate. This differs from the western blot because protein samples are not separated electrophoretically. This is then followed by detection by either nucleotide probes (for a northern blot and southern blot) or antibodies (for a western blot).
The technique offers significant savings in time, as chromatography or gel electrophoresis, and the complex blotting procedures for the gel are not required. However, it offers no information on the size of the target biomolecule. Furthermore, if two molecules of different sizes are detected, they will still appear as a single dot. Dot blots therefore can only confirm the presence or absence of a biomolecule or biomolecules which can be detected by the DNA probes or the antibody.
A radioactive sample can be hybridized to allow for the detection of variation between samples. The DNA is quantified and equal amounts are aliquoted into tubes in excess of the number of its targets in the samples, such as 10 µg for a plasmid and 1 µg for a PCR amplicon. These are denatured (NaOH and 95°C) and added to the wells where a vacuum sucks the water (with NaOH and NH4OAc) from underneath the membrane (nylon or nitrocellulose).
The sensitive dot blot test can be used to detect the Chlamydia trachomatis infection and other sexually transmitted diseases. Dot blot is used to detect Antidiacyltrehalose Antibodies in Tuberculous patients and Typhoid Fever. This test could increase the number of lives saved that are affected with these diseases. Using the dot blot test can be useful for under-developed countries especially. The dot blot is a good positive predictor of these diseases for countries and regions lacking in medical facilities and laboratories. By using this test many lives could be saved and a cure could be found for the types of diseases this test can detect.
AUTORADIOGRAPHY
Radiography is the visualisation of the pattern of distribution of radiation. In general, the radiation consists of X-rays, gamma (g ) or beta (b ) rays, and the recording medium is a photographic film. For classical X-rays, the specimen to be examined is placed between the source of radiation and the film, and the absorption and scattering of radiation by the specimen produces its image on the film. In contrast, in autoradiography the specimen itself is the source of the radiation, which originates from radioactive material incorporated into it. The recording medium which makes visible the resultant image is usually, though not always, photographic emulsion.
Autoradiography differs from the pulse-counting techniques in several ways. Each crystal of silver halide in the photographic emulsion is an independent detector, insulated from the rest of the emulsion by a capsule of gelatin. Each crystal responds to the charged particle by the formation of a latent (hidden) image that is made permanent by the process of development. The record provided by the photographic emulsion is cumulative and spatially accurate. It provides information on the localisation and distribution of radioactivity within a sample (i & ii do not do this). Thus there is little point on doing autoradiography on a specimen that is homogeneously labelled. Although it can be quantitative, autoradiography is a much slower and more difficult approach.
Nuclear emulsions have a very high efficiency for b particles (electrons of nuclear origin), particularly those with low energies. Many of the isotopes of interest to biologists have suitable isotopes, e.g. tritium (= hydrogen-3), carbon-14, , sulphur-35 and iodine-125. The effective volume of the detector emulsion in the immediate vicinity of the source may be as little as 100 cubic microns.
Autoradiography Method
- Living cells are briefly exposed to a ‘pulse’ of a specific radioactive compound.
- The tissue is left for a variable time.
- Samples are taken, fixed, and processed for light or electron microscopy.
- Sections are cut and overlaid with a thin film of photographic emulsion.
- Left in the dark for days or weeks (while the radioisotope decays). This exposure time depends on the activity of the isotope, the temperature and the background radiation (this will produce with time a contaminating increase in ‘background’ silver grains in the film).
- The photographic emulsion is developed (as for conventional photography).
- Counterstaining e.g. with toluidine blue, shows the histological details of the tissue. The staining must be able to penetrate, but not have an adverse affect on the emulsion.
- Alternatively, pre-staining of the entire block of tissue can be done (e.g. with Osmium on plastic sections coated with stripping film [or dipping emulsion] as in papers by McGeachie and Grounds) before exposure to the photographic emulsion. This avoids the need for individually (post-) staining each slide.
- It is not necessary to coverslip these slides
- The position of the silver grains in the sample is observed by light or electron microscopy Note: the grains are in a different plane of focus in the emulsion overlying the tissue section. Often oil with a x100 objective is used for detailed observation with the light microscope.
- These autoradiographs provide a permanent record.
- Full details on the batch of emulsion used, dates, exposure time and conditions should be kept for each experiment.
Maxam-Gilbert sequencing
Maxam-Gilbert sequencing is a method of DNA sequencing developed by Allan Maxam and Walter Gilbert in 1976-1977. This method is based on nucleobase-specific partial chemical modification of DNA and subsequent cleavage of the DNA backbone at sites adjacent to the the modified nucleotides.[1]
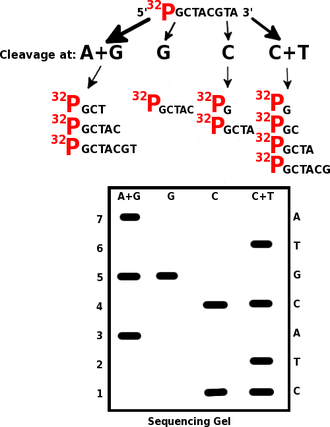
An example Maxam-Gilbert sequencing reaction. Cleaving the same tagged segment of DNA at different points yields tagged fragments of different sizes. The fragments may then be separated by gel electrophoresis.
Maxam-Gilbert sequencing was the first widely-adopted method for DNA sequencing, and, along with the Sanger dideoxy method, represents the first generation of DNA sequencing methods. Maxam-Gilbert sequencing is no longer in widespread use, having been supplanted by next-generation sequencing methods.
Procedure
Maxam-Gilbert sequencing requires radioactive labeling at one 5' end of the DNA fragment to be sequenced (typically by a kinase reaction using gamma-32P ATP) and purification of the DNA. Chemical treatment generates breaks at a small proportion of one or two of the four nucleotide bases in each of four reactions (G, A+G, C, C+T). For example, the purines (A+G) are depurinated using formic acid, the guanines (and to some extent the adenines) are methylated by dimethyl sulfate, and the pyrimidines (C+T) are methylated using hydrazine. The addition of salt (sodium chloride) to the hydrazine reaction inhibits the reaction of Thymine for the C-only reaction. The modified DNAs may then be cleaved by hot piperidine at the position of the modified base. The concentration of the modifying chemicals is controlled to introduce on average one modification per DNA molecule. Thus a series of labeled fragments is generated, from the radiolabeled end to the first "cut" site in each molecule.
The fragments in the four reactions are electrophoresed side by side in denaturing acrylamide gels for size separation. To visualize the fragments, the gel is exposed to X-ray film for autoradiography, yielding a series of dark bands each corresponding to a radiolabeled DNA fragment, from which the sequence may be inferred.[5][1]
No comments:
Post a Comment
Note: Only a member of this blog may post a comment.